Questions
- How can we Calculate the Tree Confidence?
- In phylogenetic analysis, tree confidence refers to the degree of support for a particular branch or clade in the phylogenetic tree.
There are several methods for calculating tree confidence, but some of the most common ones are:- Bootstrap Support: Bootstrap support is a non-parametric resampling method that involves generating multiple replicate data sets by sampling with replacement from the original alignment, and then analyzing each replicate data set using the same phylogenetic method.
The resulting trees are then used to calculate a support value for each branch or clade in the original tree, which reflects the proportion of the bootstrap trees that support that particular branch or clade.
Higher bootstrap support values (e.g., 70-100%) indicate stronger support for a particular branch or clade. - Bayesian Posterior Probabilities: Bayesian phylogenetic analysis uses a probabilistic model to estimate the posterior probability of each branch or clade in the tree, given the data and prior knowledge.
The posterior probabilities reflect the degree of confidence in each branch or clade, and are typically reported as a value between 0 and 1.
Higher posterior probabilities (e.g., >0.95) indicate stronger support for a particular branch or clade. - Likelihood Ratio Tests: Likelihood ratio tests are used to compare the likelihood of different tree topologies under a particular phylogenetic model, and determine whether a particular branch or clade has significantly higher likelihood than alternative hypotheses.
The test statistic is calculated as the difference in log-likelihood between the two models, and is compared to a chi-square distribution to determine the significance level.
Higher likelihood ratio values (e.g., >10) indicate stronger support for a particular branch or clade. - Jackknife Resampling: Jackknife resampling is similar to bootstrap support, but involves systematically removing a subset of the alignment (e.g., 10-20%) and analyzing the remaining data using the same phylogenetic method.
The resulting trees are then used to calculate a support value for each branch or clade in the original tree, which reflects the proportion of the jackknife trees that support that particular branch or clade.
Higher jackknife support values (e.g., >70%) indicate stronger support for a particular branch or clade.
- Bootstrap Support: Bootstrap support is a non-parametric resampling method that involves generating multiple replicate data sets by sampling with replacement from the original alignment, and then analyzing each replicate data set using the same phylogenetic method.
- In general, it is important to use multiple methods for assessing tree confidence, and to interpret the results in light of the specific phylogenetic model and data set being analyzed.
It is also important to be cautious in interpreting low support values, as they may indicate a lack of resolution or uncertainty in the data, rather than actual conflict among different hypotheses.
- In phylogenetic analysis, tree confidence refers to the degree of support for a particular branch or clade in the phylogenetic tree.
- What is Bootstrapping?
- Bootstrapping is a statistical method used to assess the reliability and robustness of a phylogenetic tree or any other statistical estimate.
In the context of phylogenetics, bootstrapping involves resampling the original sequence alignment or data set to create multiple replicate data sets, each with the same number of characters as the original data set, but with replacement of some of the characters.
Each of these replicate data sets is then analyzed using the same phylogenetic method, resulting in a set of bootstrap trees.
The bootstrap values, also known as support values, at each node of the tree are then calculated as the proportion of bootstrap trees that support the particular node or clade. - The bootstrap method is based on the principle that, given a sufficiently large sample size, the distribution of sample statistics will approach the true population distribution.
By resampling the original data set, bootstrapping provides an estimate of the variation and uncertainty in the tree topology and branch lengths.
The bootstrap values can be used to assess the strength of support for individual nodes in the tree, with higher values indicating stronger support.
A common threshold for strong support is 70-80% bootstrap value. - Bootstrapping is widely used in phylogenetic analysis to assess the reliability and robustness of phylogenetic trees, and can help to identify regions of the tree that are well supported, as well as regions that are more uncertain or ambiguous.
Bootstrapping is particularly useful for complex data sets or phylogenies, where the true relationships among taxa may be difficult to discern, and can help to identify potential sources of error or bias in the analysis.
- Bootstrapping is a statistical method used to assess the reliability and robustness of a phylogenetic tree or any other statistical estimate.
—————————————————————
IMPORTANTE Calculating Tree Confidence: BOOTSTRAPPING
In this case, the somenthing we want to calculate is the confidence of the inferred tree:
Online resource: Youtube
IMPORTANTE Calculating Tree Confidence: PARAMETRIC TESTS #TODO Formula for variance:

—————————————————————
Slides with Notes


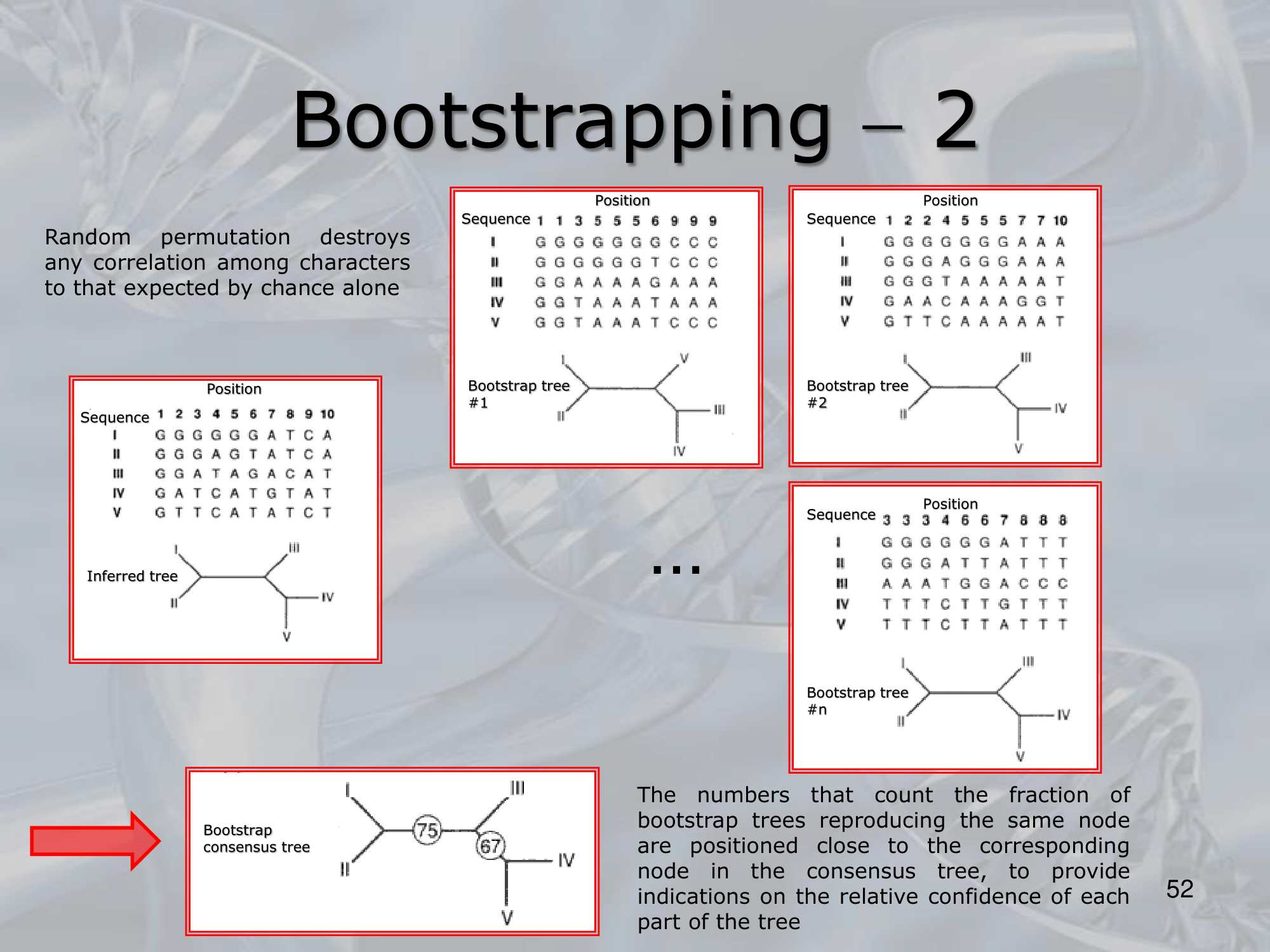
- How are the Tree Permutation Created, in the examples the GGGGGG in the Sequence I is always maintained, why is that?
- What do 75% and 67% mean in the last image?
- NOTE: Usuallly the percentage are only given on intern-branches (branches that are connected to nodes that do not exist, ancenstor specis), in this case as we can see, 75% is connected to 2 ancestor nodes, and so is 67%, all the other branches are connected to at least one node that represents a species that we know the full DNA sequence
- In all the created trees (using random permutation) 75% had the common ancestor of I and II be the common ancestor of III and 67% of those trees had the relation of the ancestor of III with the common ancestor of IV and V

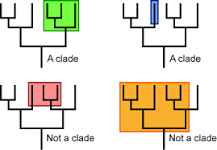
- ***What does “clade” mean?
- A group of organisms believed to comprise all the evolutionary descendants of a common ancestor.
- A clade is a grouping that includes a common ancestor and all the descendants (living and extinct) of that ancestor
- ~Example:




NOTE: We take into consideration, just 2 trees, and , we calculate (and ) which is the distance of the informative site of and the informative site of . We can also calculate the variance on more than 2 trees, but we still need to calculate and from 2 trees, it is a distance. Also we must take into consideration only the most parsimonius trees.
