Questions
- What are Outgroup and Ingroup?
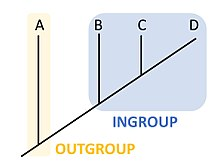
- ==In phylogenetics, an outgroup is a group of organisms that is closely related to the group of interest (called the ingroup) but is not a part of it==.
The outgroup serves as a reference point for comparing the characteristics and relationships of the organisms within the ingroup. It allows us to determine which traits are unique to the ingroup and which are shared with the outgroup. - ==For example, if we are studying the evolutionary relationships of birds, the reptiles could be an outgroup, as birds are thought to have evolved from a group of reptiles==.
By comparing the traits of birds with those of reptiles, we can identify which traits are unique to birds and which are shared with their reptilian ancestors.
- ==In phylogenetics, an outgroup is a group of organisms that is closely related to the group of interest (called the ingroup) but is not a part of it==.
- What is the Transformed Distance Method?
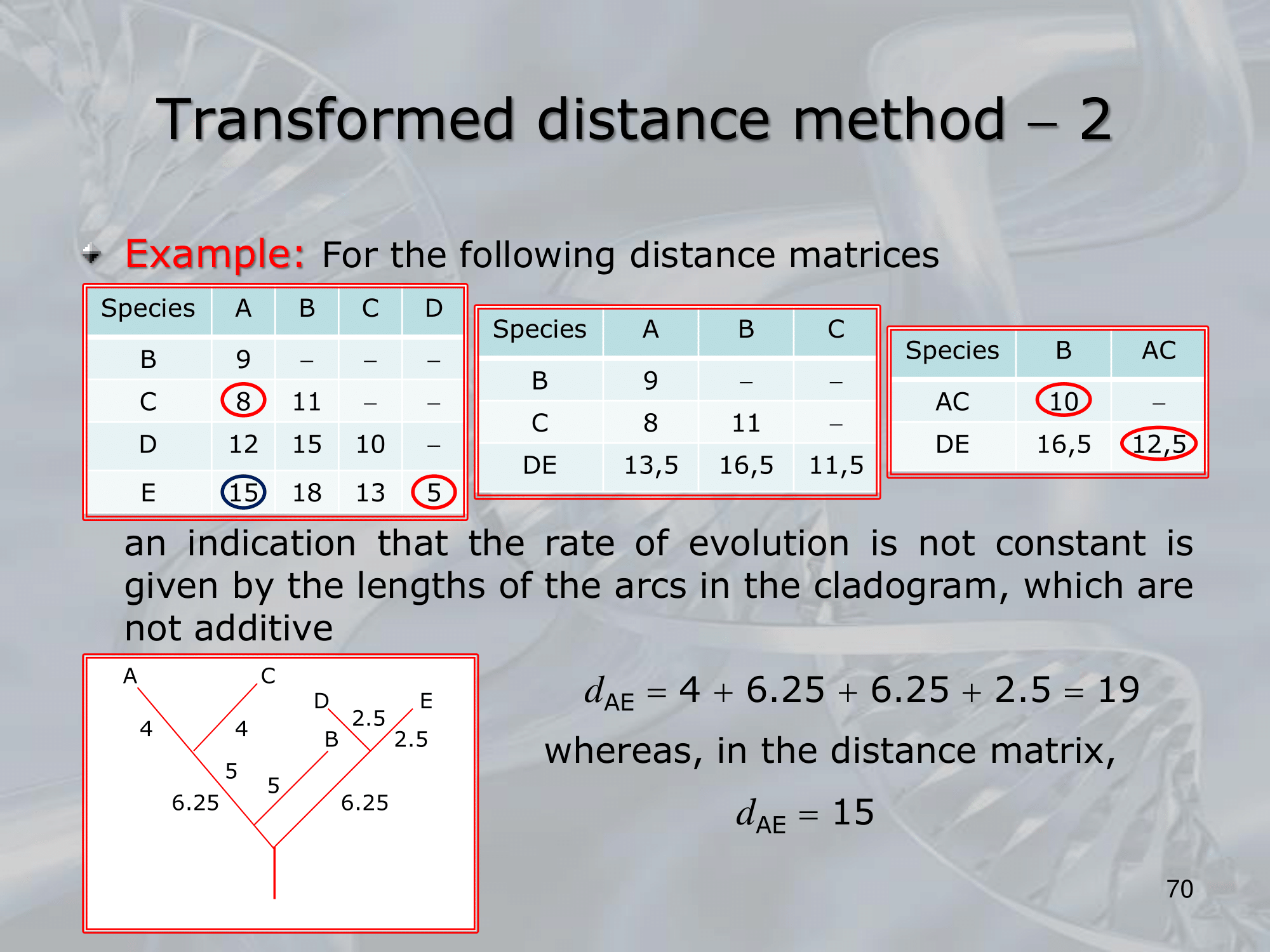
- The Transformed Distance Method is a distance-based method used for reconstructing phylogenetic trees.
It involves transforming the original distance matrix into a new matrix that satisfies certain mathematical properties, such as additivity and ultrametricity, which are necessary for constructing a valid phylogenetic tree.
The method was developed to overcome some of the limitations of traditional distance-based methods, which do not always produce accurate trees, especially when the data violate the assumptions of the model.
The Transformed Distance Method has been shown to be more robust to violations of the model assumptions and can produce more accurate trees than traditional distance-based methods.
- The Transformed Distance Method is a distance-based method used for reconstructing phylogenetic trees.
—————————————————————
IMPORTANTE
IMPORTANTE In cladistics or phylogenetics, an outgroup is a more distantly related group of organisms that serves as a reference group when determining the evolutionary relationships of the ingroup
IMPORTANTE Il Transformed distance method basato sull’UPGMA algorithm fa in modo che alla fine dei conti l’albero filogenetico uscente sia ultrametrico (ovvero che non ci siano “shortcut” tra gli archi) Partiamo da una matrice UPGMA
La distanza più corta è tra ab (17) ⇒ costruiamo i primi archi:
Ed aggiorniamo la matrice, usando le seguenti formule:
Usando come esempio la prima formula abbiamo: ⇒ La risultante matrice è:
Ripetiamo l’algoritmo, la nuova distanza minima è 22 tra (ab)e, il ramo che aggiungiamo dovrà tenere conto del precedente (di lunghezza ), quindi:
La matrice va riaggiornata, e le formule questa volta saranno:
(Nota che adesso si fa una media su 3 unità taxonomiche, e che le distanze che si usano sono sempre quelli della matrice iniziale) Oppure possiamo usare le distanze della matrice precedente cambiando leggermente la formula, ottenendo lo stesso risultato:
La matrice risultante sarà:
Distanza minore di cd
Usando la matrice precedente per il calcolo delle nuove distanze:
Or we can use the other formula: Dove: Il risultato è lo stesso:
L’albero finale sarà: (((a,b,),e),(c,d))
Online resource: Youtube
La formula che utilizziamo per creare la “giusta” distanza dei rami è: In caso di nodi appartenenti ad allo stesso “mega-cluster” ~Es.: In caso di nodi appartenenti a due “mega-cluster” diversi ~Es.: In questo caso i due “mega-cluster” sono &
—————————————————————
Slides with Notes


IMPORTANTE In cladistics or phylogenetics, an outgroup is a more distantly related group of organisms that serves as a reference group when determining the evolutionary relationships of the ingroup





IMPORTANTE Il Transformed distance method basato sull’UPGMA algorithm fa in modo che alla fine dei conti l’albero filogenetico uscente sia ultrametrico (ovvero che non ci siano “shortcut” tra gli archi) Partiamo da una matrice UPGMA
L’albero finale sarà: (((a,b,),e),(c,d))