Questions
- What are Simple Alignments?
- Simple alignments are a type of sequence alignment in bioinformatics that involves aligning two sequences based on their similarities and differences. In simple alignments, the goal is to maximize the number of matching nucleotides or amino acids between the two sequences while minimizing the number of gaps inserted to align them.
- Simple alignments are usually performed using dynamic programming algorithms, such as the Needleman-Wunsch algorithm or the Smith-Waterman algorithm.
These algorithms generate a matrix of scores that represents the alignment between the two sequences, where each cell in the matrix corresponds to a pair of nucleotides or amino acids in the sequences.
The scores in the matrix are used to determine the optimal alignment between the two sequences. - Simple alignments are used in a variety of applications in bioinformatics, such as identifying homologous genes, predicting protein structures, and studying evolutionary relationships between organisms.
- Can you make an Example of Simple Alignment?
- Here is an example of a simple alignment between two DNA sequences:
- Sequence 1: ATCGATCGATCGATCGATCGATCG
- Sequence 2: ATCGATCGAGCGATCGATCGACCG
- To align these sequences, we can use a technique called pairwise alignment. The goal of pairwise alignment is to find the best possible alignment between two sequences, based on a scoring system that takes into account the similarity between the nucleotides or amino acids.
- One commonly used scoring system is the BLOSUM matrix, which assigns scores based on the frequency of each amino acid substitution in a large dataset of related proteins.
- Using a simple scoring system, such as matching nucleotides are worth 1 point and mismatches are worth -1 point, we can generate the following alignment:
- ATCGATCGATCGATCGATCGATCG
- ATCGATCGAGCGATCGATCGACCG
- In this alignment, the matching nucleotides are aligned vertically, and gaps are inserted to account for the differences between the two sequences.
- Note that this is just one possible alignment, and there are often multiple ways to align two sequences depending on the scoring system used and the specific algorithm implemented.
- Here is an example of a simple alignment between two DNA sequences:
- What are the types of Mutations that can happen on the DNA?
- There are several types of mutations that can occur on the DNA, including:
- ==Point mutations: This type of mutation involves the substitution of a single nucleotide with another nucleotide. There are three types of point mutations==:
- ==Silent mutations: These mutations do not result in a change in the amino acid sequence of the protein because the substituted nucleotide codes for the same amino acid as the original nucleotide==.
- ==Missense mutations: These mutations result in a change in the amino acid sequence of the protein because the substituted nucleotide codes for a different amino acid than the original nucleotide==.
- ==Nonsense mutations: These mutations result in the premature termination of the protein synthesis because the substituted nucleotide creates a stop codon where there should not be one==.
- ==Insertions and Deletions: These mutations involve the addition or removal of one or more nucleotides from the DNA sequence==.
These mutations can cause frameshifts, where the reading frame of the genetic code is altered, leading to a completely different protein sequence. - ==Inversions: This type of mutation involves the reversal of the order of a segment of DNA==.
- Translocations: This type of mutation involves the movement of a segment of DNA from one location to another in the genome. This can lead to changes in gene expression and function.
- Duplications: This type of mutation involves the duplication of a segment of DNA, leading to multiple copies of a particular gene or genes. This can also cause changes in gene expression and function.
- ==Point mutations: This type of mutation involves the substitution of a single nucleotide with another nucleotide. There are three types of point mutations==:
- These mutations can occur spontaneously or be induced by environmental factors such as radiation or chemicals, and they can have various effects on the organism, ranging from no effect to severe disease or even death.
- There are several types of mutations that can occur on the DNA, including:
—————————————————————
IMPORTANTE
IMPORTANTE L’allineamento di due sequenze di nucleotidi o amminoacidi può essere un indicatore della loro relazione evolutiva, detta omologia, ma purtroppo non possiamo dare uno “score” all’emologia.
IMPORTANTE In ogni punto di una sequenza di basi possano accadere uno di 3 mutazioni:
- Sostituzione: una base cambia in un’altra, l’evento più probabile tra i tre.
- Inserzione: vengono aggiunte una o più basi tra due già esistenti.
- Cancellazione: vengono rimosse una o più basi.
NOTA: La sostituzione è molto più probabile delle altre due mutazioni. In caso accada un’inserzione o una cancellazione, è probabile che essa aggiunga/tolga più di una singola base.
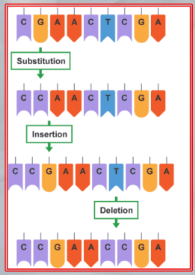
IMPORTANTE Per valutare quale tra tutti i possibili allineamenti di due sequenze (di misura diversa) è “l’ottimale”, possiamo usare il metodo di “Simple Allignments”. ~Ex.:
Diamo un punteggio sulla base di: : se due basi sono identiche. : se due basi sono diverse. Nell’esempio i tre allineamenti hanno uno score di 4, 1, 3.
~Ex.: Consideriamo la possibilità di “Gaps”. Il Gap sta a significare che in quel punto è avvenuto un “fenomento indel” (un’inserzione od una cancellazione.)
Le possibilità aumentano esponenzialmente, da 3 possibili combinazioni passiamo a 28.
—————————————————————
Slides with Notes





IMPORTANTE L’allineamento di due sequenze di nucleotidi o amminoacidi può essere un indicatore della loro relazione evolutiva, detta omologia, ma purtroppo non possiamo dare uno “score” all’emologia.
IMPORTANTE In ogni punto di una sequenza di basi possano accadere uno di 3 mutazioni:
- Sostituzione: una base cambia in un’altra, l’evento più probabile tra i tre.
- Inserzione: vengono aggiunte una o più basi tra due già esistenti.
- Cancellazione: vengono rimosse una o più basi.
NOTA: La sostituzione è molto più probabile delle altre due mutazioni. In caso accada un’inserzione o una cancellazione, è probabile che essa aggiunga/tolga più di una singola base.
IMPORTANTE Per valutare quale tra tutti i possibili allineamenti di due sequenze (di misura diversa) è “l’ottimale”, possiamo usare il metodo di “Simple Allignments”. ~Ex.:
~Ex.: Consideriamo la possibilità di “Gaps”. Il Gap sta a significare che in quel punto è avvenuto un “fenomento indel” (un’inserzione od una cancellazione.)