Questions
- How can we Correctly Estimate the Arc Length in Phylogenetic Trees?
- To correctly estimate the arc length in phylogenetic trees, one needs to consider the branch lengths and the topology of the tree.
The arc length is the distance between two nodes on a tree, which is equivalent to the number of substitutions that have occurred along the evolutionary path between the two nodes. - To estimate the arc length, one can use various methods, such as the maximum likelihood and Bayesian approaches.
These methods take into account the branch lengths, the substitution model used, and the variability in the substitution rates across different lineages.
The maximum likelihood approach estimates the arc length by finding the branch lengths that maximize the likelihood of the observed data, while the Bayesian approach estimates the posterior probability distribution of the arc length given the data. - It is important to note that the estimation of the arc length can be affected by several factors, such as the quality and completeness of the data, the choice of the substitution model, and the level of divergence between the sequences being compared.
Therefore, it is advisable to use multiple methods and evaluate the robustness of the results to different assumptions and parameter settings.
- To correctly estimate the arc length in phylogenetic trees, one needs to consider the branch lengths and the topology of the tree.
- What are Cladograms?
- A cladogram is a type of phylogenetic tree that represents the evolutionary relationships among different taxa or groups of organisms.
Cladograms show the branching pattern of evolutionary relationships based on shared derived characteristics or traits (called synapomorphies) among taxa.
In other words, a cladogram groups organisms based on their shared features, rather than on overall similarity. - In a cladogram, each branch point or node represents the hypothetical common ancestor of the taxa that emerge from it.
The length of the branches does not necessarily reflect the evolutionary distance between taxa, but rather their relative divergence from their common ancestor.
Cladograms can be constructed using different types of molecular data, morphological data, or a combination of both. - Cladistics is a method of classification based on the analysis of shared characteristics among taxa, and cladograms are the visual representations of the results of such analyses.
Cladograms are widely used in biology to study the evolutionary relationships among organisms and to test hypotheses about their evolutionary history.
- A cladogram is a type of phylogenetic tree that represents the evolutionary relationships among different taxa or groups of organisms.
—————————————————————
IMPORTANTE
IMPORTANTE Un problema con l’algoritmo UPGMA sono i risultanti pesi sugli archi:
In questo esempio abbiamo che tra A ed E c’è una differenza diversa da 0 dal loro antenato comune, il che non dovrebbe essere possibile.
—————————————————————
Slides with Notes

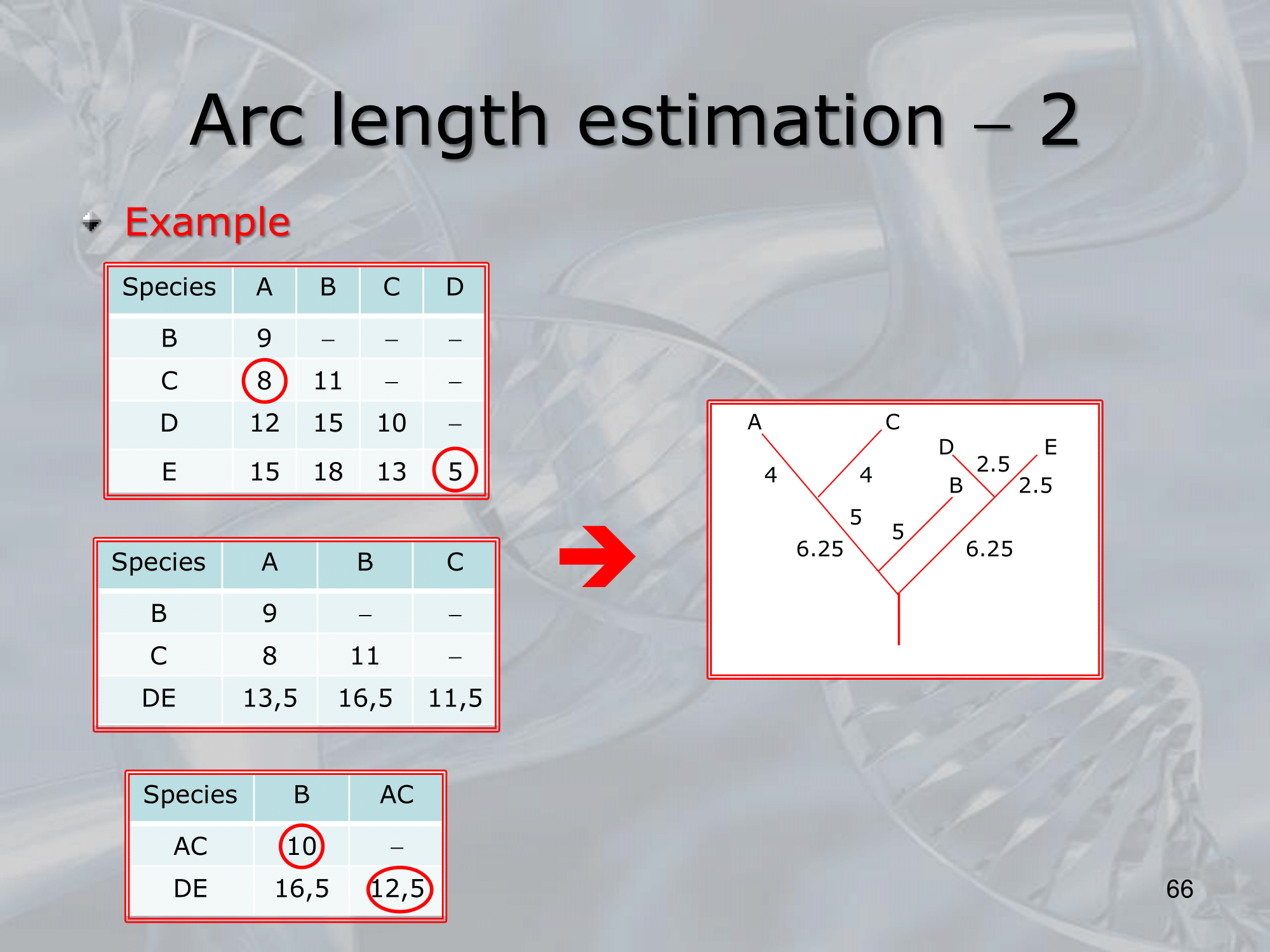


IMPORTANTE Un problema con l’algoritmo UPGMA sono i risultanti pesi sugli archi: