Questions
- How can we Estimate the Substitution Number?
- The substitution number, or the number of substitutions that have occurred between two DNA or protein sequences, can be estimated using various methods in molecular evolution, such as maximum likelihood, Bayesian inference, and parsimony methods.
- ==Maximum likelihood and Bayesian inference methods use a statistical model of evolution to estimate the most likely number of substitutions that have occurred between two sequences==.
These methods consider the frequency of nucleotide or amino acid substitutions, as well as the patterns of substitution that occur at different sites in the sequence. - ==Parsimony methods, on the other hand, rely on the principle of Occam’s razor to choose the simplest explanation for the observed differences between two sequences==.
This method counts the number of changes that are required to explain the observed differences between the two sequences and estimates the substitution number based on the minimum number of changes.
- ==Maximum likelihood and Bayesian inference methods use a statistical model of evolution to estimate the most likely number of substitutions that have occurred between two sequences==.
- Overall, estimating the substitution number between two sequences is important for understanding their evolutionary relationship and the degree of divergence between them.
- The substitution number, or the number of substitutions that have occurred between two DNA or protein sequences, can be estimated using various methods in molecular evolution, such as maximum likelihood, Bayesian inference, and parsimony methods.
- What is the James-Cantor Model?
- The James-Cantor model is a mathematical model used in molecular evolution to estimate the number of nucleotide substitutions that have occurred between two homologous DNA or RNA sequences.
It assumes that the nucleotide substitution rate is constant over time and that all nucleotide substitutions are equally likely.
The model takes into account the possibility of multiple substitutions at the same site (i.e., parallel substitutions) and incorporates the observed frequencies of the four nucleotides in the two sequences being compared.
The model can be used to estimate the number of synonymous and non-synonymous substitutions separately, which can be used to study the evolution of protein-coding genes.
The James-Cantor model is one of the simplest nucleotide substitution models and is widely used in phylogenetic analysis and molecular clock studies.
- The James-Cantor model is a mathematical model used in molecular evolution to estimate the number of nucleotide substitutions that have occurred between two homologous DNA or RNA sequences.
- How did the James-Cantor Model Evolve?
- The James-Cantor Model is a mathematical model that describes the evolution of DNA sequences over time. It was first proposed by James and Cantor in 1975 and has since undergone several modifications and improvements
- The original James-Cantor Model assumed that nucleotide substitutions occurred independently at each site in the DNA sequence, and that all four nucleotides had equal probability of being substituted.
However, subsequent research has shown that the actual process of nucleotide substitution is more complex and depends on factors such as the chemical properties of the nucleotides and their neighboring bases. - Therefore, several modifications and extensions to the James-Cantor Model have been proposed, including more sophisticated substitution models that take into account the effects of neighboring nucleotides, as well as models that account for different rates of evolution at different sites in the DNA sequence.
These models have been used extensively in molecular evolution research to estimate evolutionary relationships and divergence times between species, and to identify sites in DNA sequences that are under positive or negative selection.
- Can you Explain the Math behind the James-Cantor Model?
- The James-Cantor model is a probabilistic model that calculates the expected number of substitutions between two DNA sequences over a given time period. The basic assumptions of the model are that the substitution rates are equal among sites in the sequence and that each site evolves independently. The model also assumes that the nucleotide frequencies are constant over time and that all substitutions are equally likely.
- The model is based on the use of a rate matrix, which describes the probability of a nucleotide substitution from one base to another. The rate matrix is defined as follows:
Q = { q_ij }
Where:q_ijis the rate of substitution from nucleotideito nucleotidej.- The diagonal elements
q_iiare all equal to zero, since a nucleotide cannot substitute for itself.
- The off-diagonal elements are given by:
q_ij = α_ij * β_j
Where:α_ijis the probability of a nucleotide substitution fromitojandβ_jis the frequency of nucleotidej.
- The expected number of substitutions between two sequences is then calculated using the following formula:
d = t * S
Where:dis the expected number of substitutionstis the time in evolutionary unitsSis the number of differences between the two sequences.
The value ofSis usually estimated using methods such as pairwise alignment or multiple sequence alignment.
- The James-Cantor model can be extended to include more complex evolutionary scenarios, such as changes in the substitution rates over time or among different lineages. However, the basic model provides a useful framework for understanding the evolution of DNA sequences and for estimating the amount of evolutionary change between different sequences.
—————————————————————
IMPORTANTE
IMPORTANTE James-Cantor Model: La possibilità che un nucelotida diventi un nucelotida diverso è pari a mentre la possibiltà che rimanga lo stesso è . Osservando due specie, il cui fenomeno di speciazione è avvenuto fa, la possibilità che continuino a portare lo stesso singolo nucelotida “C” è pari a: Si considera che il rate di replecemnt frequency di un singolo nucelotida con un altro possibile sia , allora il rate di replacement di tutti i nucleotidi è . Allora, il numero di sostituzioni sarà uguale a Per calcolare prendiamo la frazione di siti che differiscono tra le due specie: Quindi: L’equazione finale sarà:
IMPORTANTE James-Canter Model:
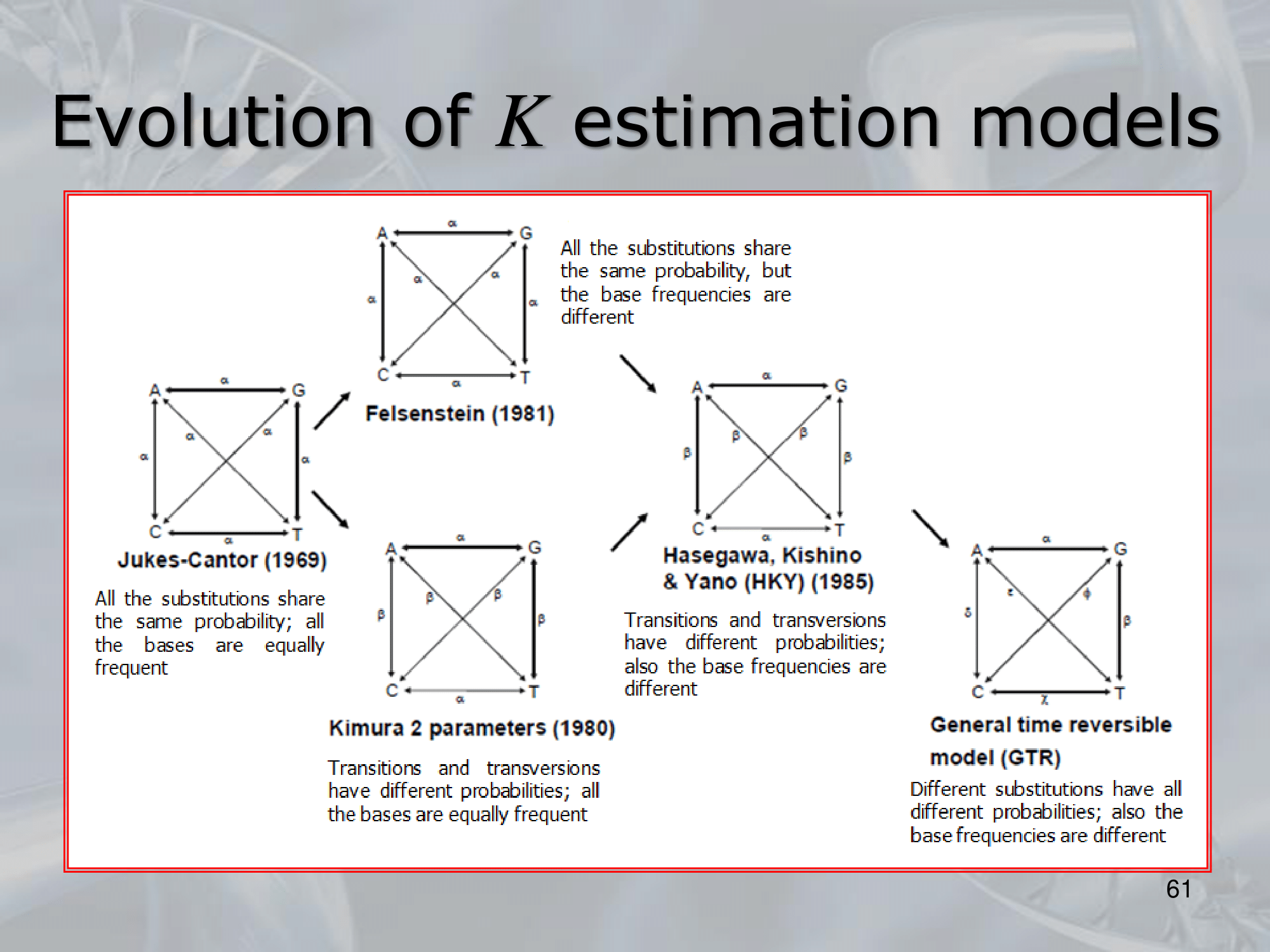
Tutte le basi hanno stesso rate di sostituzione .
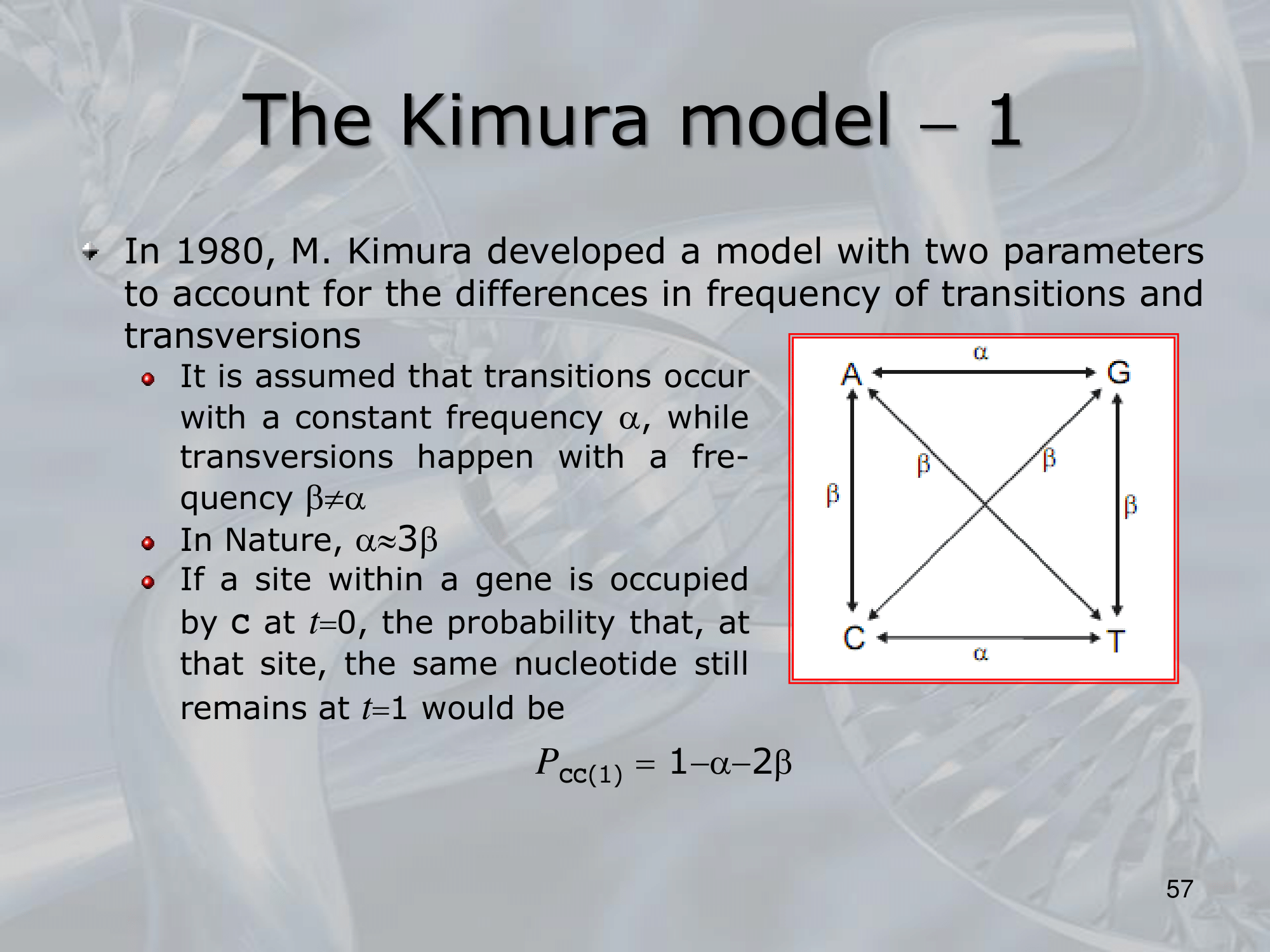
IMPORTANTE Kimura Model:
Se sostituiamo una Purina (G & A) con l’altra purina, il rate di sostituzione è , lo stesso se sostituiamo una Pirimidina (C & T) con l’altra pirimidina. Al contrario se passiamo da purina a pirimidina o viceversa, il rate di sostituzione diventa (più improbabile).
IMPORTANTE General Time Reversible Model (GTR): Tutte le sostituzioni hanno rate diversi.
—————————————————————
Slides with Notes










IMPORTANTE James-Cantor Model: La possibilità che un nucelotida diventi un nucelotida diverso è pari a mentre la possibiltà che rimanga lo stesso è . Osservando due specie, il cui fenomeno di speciazione è avvenuto fa, la possibilità che continuino a portare lo stesso singolo nucelotida “C” è pari a: Si considera che il rate di replecemnt frequency di un singolo nucelotida con un altro possibile sia , allora il rate di replacement di tutti i nucleotidi è . Allora, il numero di sostituzioni sarà uguale a Per calcolare prendiamo la frazione di siti che differiscono tra le due specie: Quindi: L’equazione finale sarà:




IMPORTANTE James-Canter Model:
IMPORTANTE Kimura Model:
IMPORTANTE General Time Reversible Model (GTR): Tutte le sostituzioni hanno rate diversi.