Questions
- What is Ligand Docking?
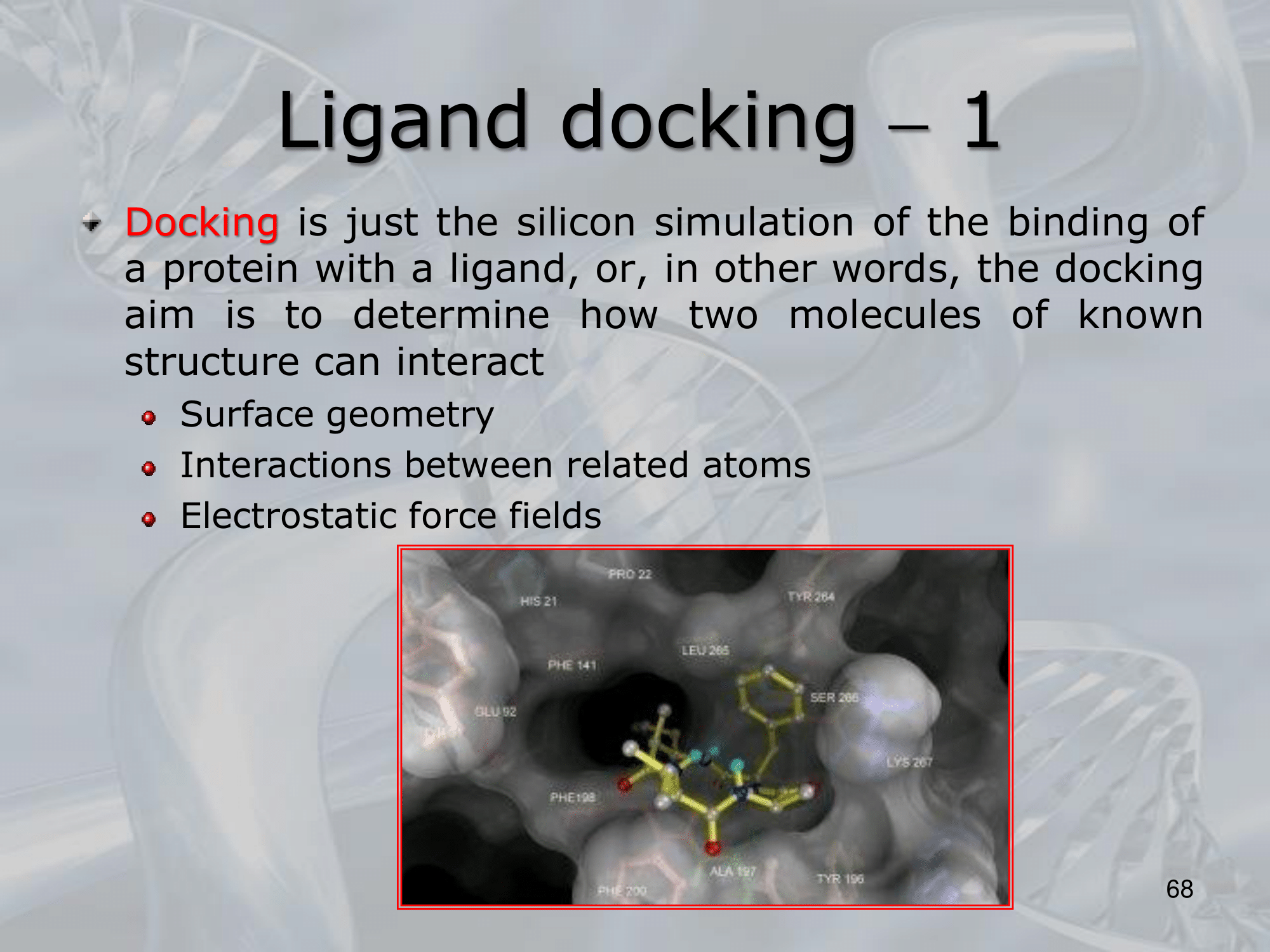
- Ligand docking is a computational technique used in drug discovery and molecular biology to predict the binding orientation and affinity of a small molecule, called a ligand, with a larger biomolecular target, such as a protein or nucleic acid.
- The goal of ligand docking is to predict the most energetically favorable binding conformation of a ligand in the binding site of a target molecule, which can help identify potential drug candidates or provide insights into the molecular mechanisms of biological processes.
- The ligand docking process typically involves three steps:
- Preparation of the target molecule, which involves the generation of a three-dimensional model of the target structure and the identification of the binding site where the ligand is expected to bind.
- Preparation of the ligand molecule, which involves the generation of a three-dimensional model of the ligand structure and the identification of any potential binding conformations.
- Docking simulation, which involves the prediction of the binding orientation and affinity of the ligand with the target molecule. This is typically done using computational algorithms that calculate the energetics and geometric complementarity of the ligand and target.
- Overall, ligand docking is an important tool in drug discovery and molecular biology, as it can help identify potential drug candidates, provide insights into the molecular mechanisms of biological processes, and guide experimental design.
- What is the Difference between Ligand Screening and Ligand Docking?
- Ligand screening and ligand docking are both computational methods used in drug discovery to identify potential small-molecule compounds that can bind to a target protein or enzyme.
However, there are differences between the two methods:
- Ligand screening: Ligand screening is a virtual screening method that involves testing a large library of compounds against a target protein to identify potential ligands that can bind to the protein.
The library of compounds can be either commercially available or generated computationally.
Ligand screening can be used to identify potential lead compounds that can be further optimized for drug development. - Ligand docking: Ligand docking is a computational method that predicts the binding mode and affinity of a small-molecule ligand to a target protein or enzyme.
Docking typically involves two main steps: first, predicting the possible binding sites on the protein or enzyme, and second, generating and scoring conformations of the ligand in the predicted binding site.
Docking can be used to prioritize compounds identified from ligand screening or to refine the binding mode and affinity of a known ligand.
- In summary, ligand screening is a method to identify potential lead compounds from a large library of compounds, while ligand docking is a method to predict the binding mode and affinity of a specific ligand to a target protein or enzyme.
Ligand screening can be used as a preliminary step to identify potential ligands, and ligand docking can be used to refine the binding mode and affinity of a ligand.
- Ligand screening and ligand docking are both computational methods used in drug discovery to identify potential small-molecule compounds that can bind to a target protein or enzyme.
—————————————————————
IMPORTANTE
IMPORTANTE In many cases the three dimensional structure a protein and of its ligands are known, what we search for using heuristic searches and suboptimal solutions is the final structure (protein + ligand)
IMPORTANTE As in protein folding, this are the main parts to this problem:
- Define an energy function, it will evalate the quality of the final structure
- Define a search algorithm, how the program will search given the space of possibilites
- Managing the flexibility of both the protein and the putative ligand: #IMPORTANTE Key-Lock Approach to Ligand Docking: We assume a rigid protein structure which binds to a ligand with a flexibile structure #IMPORTANTE Induced Fit Docking: Both the protein and the ligand are considered flexible #IMPORTANTE Compromise: Assuming a rigid protein backbone, while allowing the flexibilty of the side chains near the bindign site.
IMPORTANTE Auto-Dock method A famous method for both flexible and rigid macromolecules Initially it used the Monte-Carlo Algorithm, allowing some “bad moves” to find a more exhaustive final soltuion
Today it uses Genetic Algorithms.
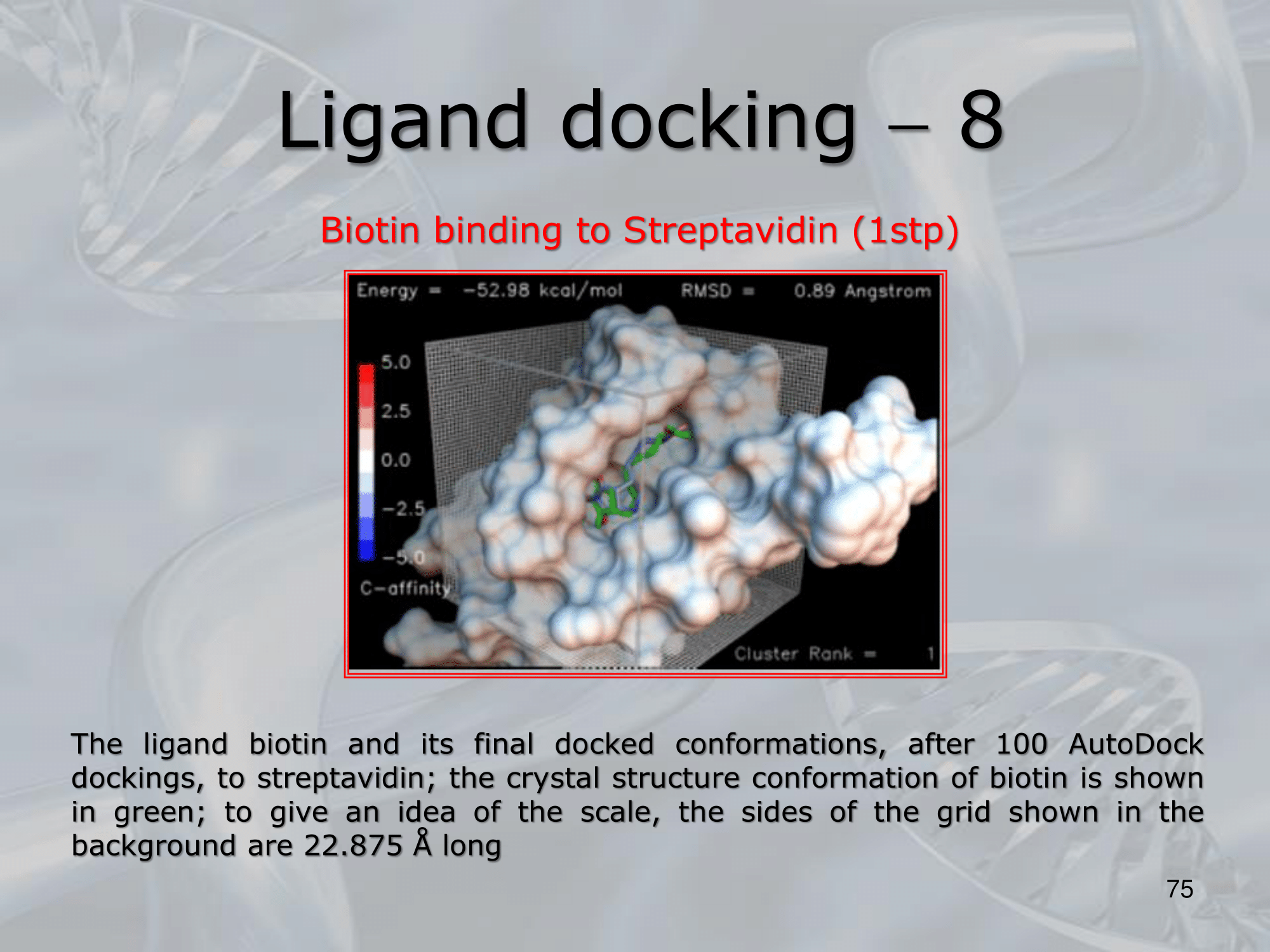
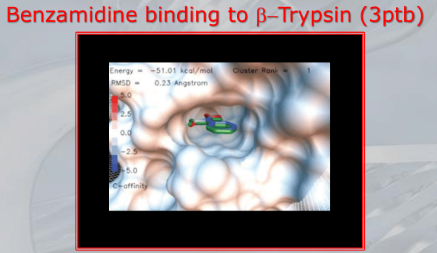
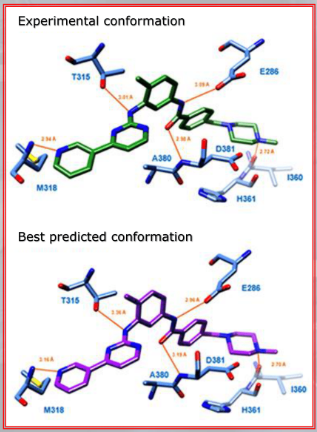
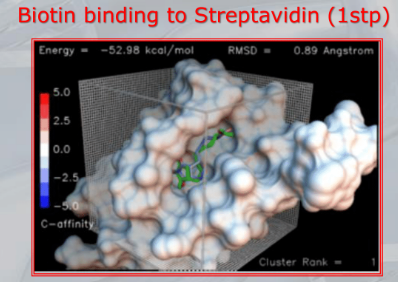
~Ex.: Auto-Docking results:
—————————————————————
Slides with Notes