Questions
- What is Threading?
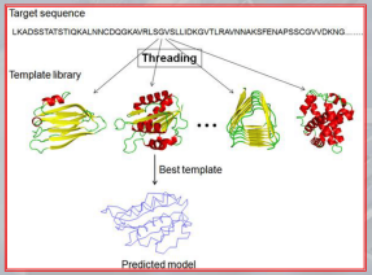
- Threading, also known as fold recognition, is a computational method for predicting the three-dimensional structure of a protein based on its amino acid sequence and a library of known protein structures.
Unlike comparative modeling, which relies on a homologous protein structure to predict the structure of a target protein, threading uses a library of known protein structures to search for possible fold templates that can be used to predict the structure of a target protein. - Threading typically involves several steps.
- First, the amino acid sequence of the target protein is compared to a library of known protein structures to identify potential templates that share sequence similarity with the target protein.
- Next, the sequence of the target protein is threaded onto the structures of the potential templates to generate a set of possible models.
- Finally, the models are evaluated using a scoring function that assesses how well the model fits the target sequence and how energetically favorable it is.
- Threading is a valuable tool for predicting the structure of proteins that do not have close homologs with known structures or for which comparative modeling is not applicable.
It is also useful for predicting the structures of proteins with novel folds that have not been observed before.
However, threading can be computationally intensive and can require a large library of known protein structures to be effective.
- Threading, also known as fold recognition, is a computational method for predicting the three-dimensional structure of a protein based on its amino acid sequence and a library of known protein structures.
- What is exactly a Protein Class?
- In protein science, a protein class is a group of proteins that share similar structural or functional characteristics.
Proteins can be classified in various ways, depending on the criteria used to group them.
For example, proteins can be classified based on their overall shape or fold, the types of secondary structural elements they contain, the types of amino acid sequences they have, or the functions they perform. - One widely used classification scheme is the Structural Classification of Proteins (SCOP), which divides proteins into a hierarchical set of classes, folds, superfamilies, and families based on their overall structural similarities.
Another classification scheme is the Enzyme Commission (EC) number system, which categorizes enzymes based on the type of reaction they catalyze and the chemical bonds involved in the reaction. - Protein classes are useful for organizing and understanding the vast diversity of proteins found in nature.
By identifying common features among proteins in a class, scientists can gain insights into the evolution and function of these molecules and develop new strategies for studying and manipulating them.
- In protein science, a protein class is a group of proteins that share similar structural or functional characteristics.
—————————————————————
IMPORTANTE
IMPORTANTE The number of protein TOPOLOGY existing in nature is finate and “small”: topopogyies. #IMPORTANTE Protein Threading: Given a 3D structure of a folded protein, to which strucutural class can it belong? #IMPORTANTE Elements of a threading system:
- A library of 3D strucures.
- An energy function.
- A threading algortihm: to compare the minimum energy allignment between the sequence and the tested structures.
- A statistical evaluation criterion.
Many databases of hierchical structure have been developt (like CATH and SCOP), and we can use them even for protein folding, given a sequence of amminoacid we can know to which class it belongs, having a base structure:
TODO What is exactly a protein class?
—————————————————————
Slides with Notes

IMPORTANTE The number of protein TOPOLOGY existing in nature is finate and “small”: topopogyies. #IMPORTANTE Protein Threading: Given a 3D structure of a folded protein, to which strucutural class can it belong? #IMPORTANTE Elements of a threading system:
- A library of 3D strucures.
- An energy function.
- A threading algortihm: to compare the minimum energy allignment between the sequence and the tested structures.
- A statistical evaluation criterion.
Many databases of hierchical structure have been developt (like CATH and SCOP), and we can use them even for protein folding, given a sequence of amminoacid we can know to which class it belongs, having a base structure:
TODO What is exactly a protein class?



