Questions
- What are Lattice Models?
- Lattice models are simplified models of protein folding that represent the protein as a chain of connected beads or points on a lattice.
In a lattice model, the protein is assumed to be constrained to move only along the lattice points, which simplifies the calculations required to model protein folding. - In a lattice model, the protein is represented by a sequence of connected lattice points, with each point representing an amino acid residue.
The interactions between adjacent lattice points are described by a simplified energy function that takes into account the local geometry of the protein chain. - Lattice models are useful for exploring the folding of small proteins and for studying the basic principles of protein folding, such as the role of hydrophobic interactions and the formation of secondary structures.
They are computationally efficient and allow for the rapid exploration of large conformational spaces. - However, lattice models also have limitations.
They are highly simplified and do not take into account the full complexity of protein folding, such as the effects of solvent interactions and the role of non-local interactions in guiding the folding process.
Therefore, while lattice models can provide insight into the basic principles of protein folding, they may not be suitable for modeling the folding of larger or more complex proteins.
- Lattice models are simplified models of protein folding that represent the protein as a chain of connected beads or points on a lattice.
—————————————————————
IMPORTANTE
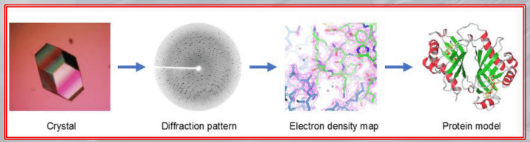
IMPORTANTE What are Lattice Models?: ==The protein folding is represnted in a 2D or 3D grid== The lattice statistical model seeks to recreate protein folding by minimizing the free energy of the contacts between hydrophobic amino acids (respecting principaly the “hydrophobic rule”). #IMPORTANTE Ground Truth: the ground truth for comparing actual protein folding, with the results from lattice models are given by the X-ray Crystallography:
IMPORTANTE H-P Model (Hydrophobic-Polar) : the simplest lattice model.
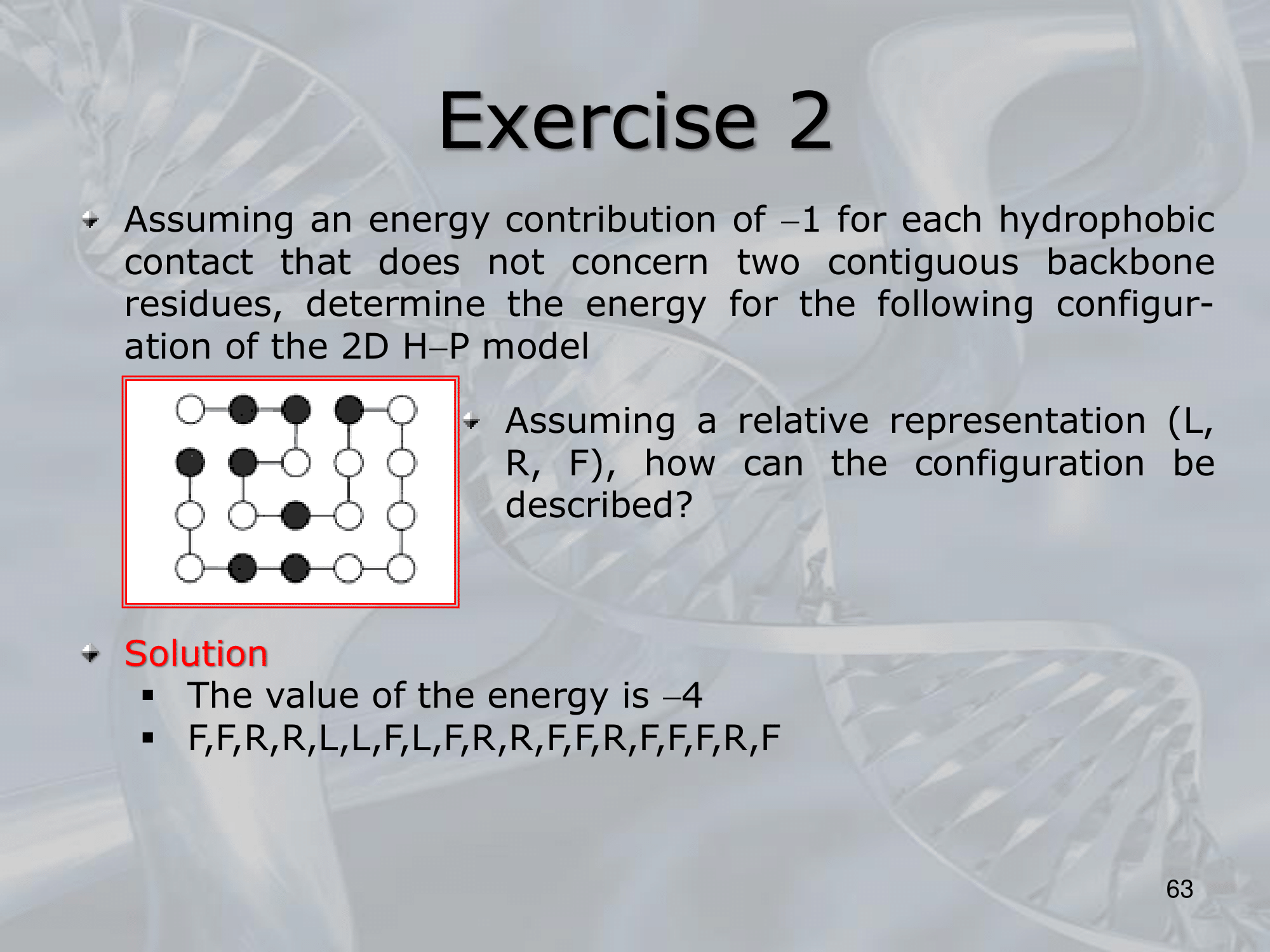
The configuration score which we wish to minimize is given by the number of hydrophobi contact in the grid (each contatct values , so the more contacts the better)
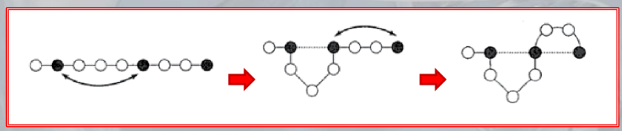
IMPORTANTE For the H-P Model it was suggested that a possible mechanism for the secondary structure to maximize the number of contacts was to form U-turns in antiparallel -sheets:
IMPORTANTE As the Levinthal Paradox states it’s impossible to try each possible combination, so soft computing models are necessary, such as: Monte Carlo methods Machine Learning Genetic Algorithms
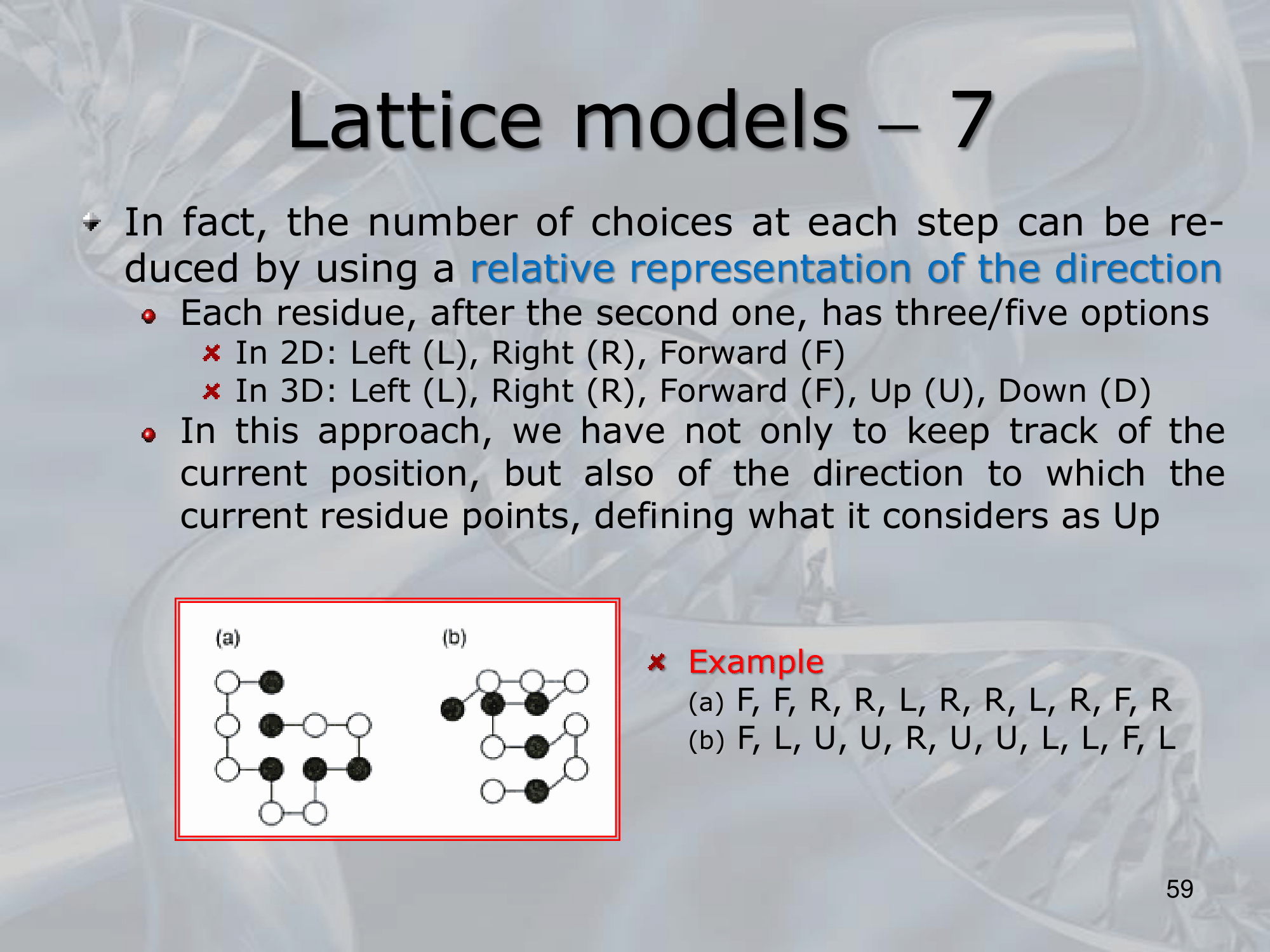
IMPORTANTE One benefit of Lattice Models is the absolute representation of the direction, using the following names: R (Right), D (down), , we can create:
Or we can use the relative representation, (relative to the previous residue):
#IMPORTANTE We need to avoid steric collision, for example if we go like L, L, L, L, in the relative representation we will return to the (0,0) starting point creating a collision of two residues (calld a steric collision). We can avoid steric collisions using an an injective method during the creation of the lattice model (this creation is also called protein embeddingNOT_SURE_ABOUT_THIS ) where we add one residue at the time avoiding a collision every time. Or we can assing a really high value to the configuration where we find a collision.
—————————————————————
Slides with Notes
IMPORTANTE What are Lattice Models?: ==The protein folding is represnted in a 2D or 3D grid== The lattice statistical model seeks to recreate protein folding by minimizing the free energy of the contacts between hydrophobic amino acids (respecting principaly the “hydrophobic rule”). #IMPORTANTE Ground Truth: the ground truth for comparing actual protein folding, with the results from lattice models are given by the X-ray Crystallography:


IMPORTANTE H-P Model (Hydrophobic-Polar) : the simplest lattice model.

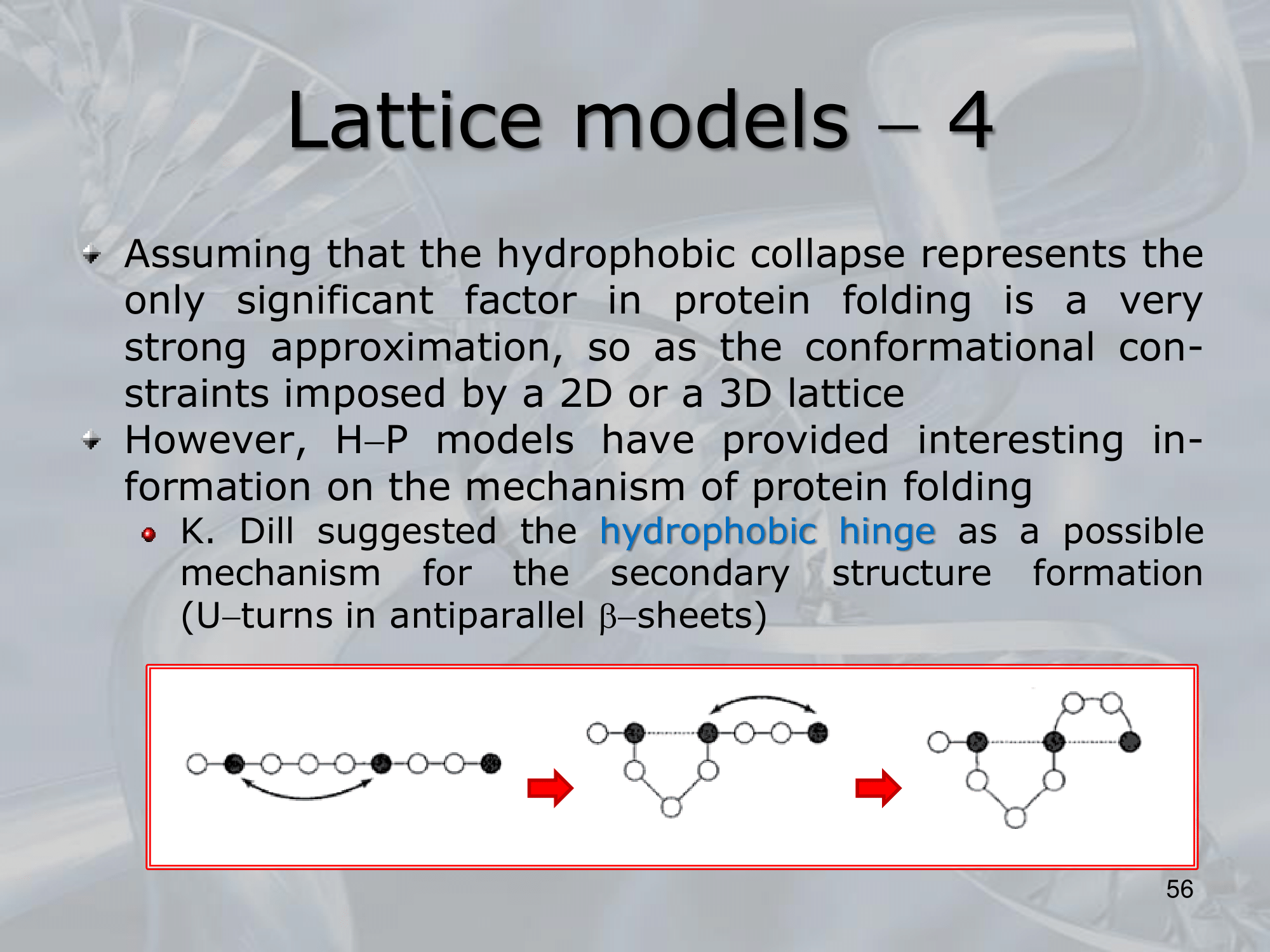
IMPORTANTE For the H-P Model it was suggested that a possible mechanism for the secondary structure to maximize the number of contacts was to form U-turns in antiparallel -sheets:

IMPORTANTE As the Levinthal Paradox states it’s impossible to try each possible combination, so soft computing models are necessary, such as: Monte Carlo methods Machine Learning Genetic Algorithms
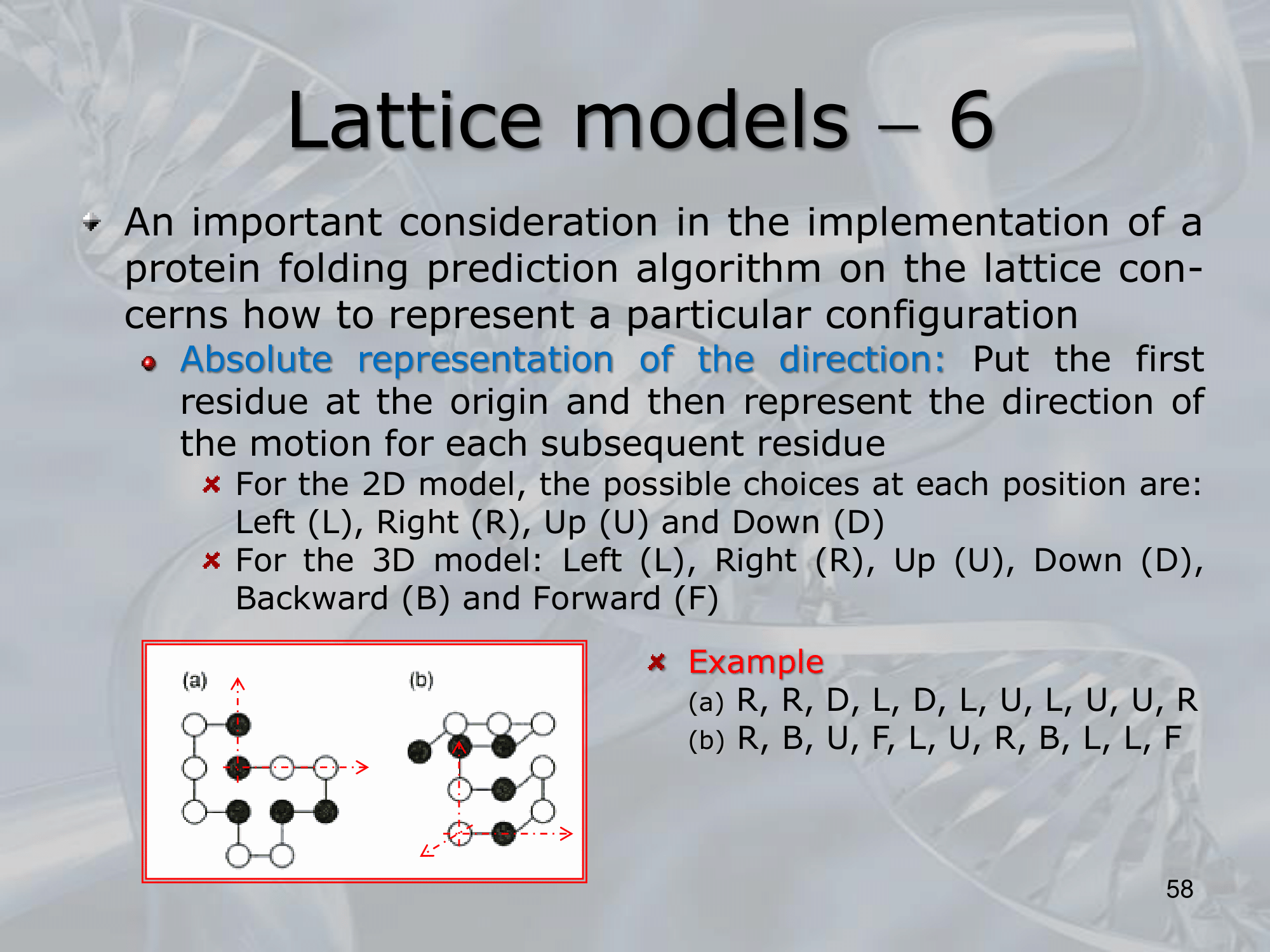
IMPORTANTE One benefit of Lattice Models is the absolute representation of the direction, using the following names: R (Right), D (down), , we can create: