Questions
- What we mean with Character-Based Molecular Data?
- Character-based molecular data refers to the use of specific genetic or protein sequences, such as nucleotide or amino acid sequences, to infer evolutionary relationships between species.
These sequences are used as “characters” to build phylogenetic trees, which can help to understand the evolutionary history of different organisms. - Character-based molecular data can be obtained through techniques such as DNA sequencing, which allow for the comparison of genetic sequences across different species.
The differences and similarities in these sequences can then be used to infer evolutionary relationships and construct phylogenetic trees.
- Character-based molecular data refers to the use of specific genetic or protein sequences, such as nucleotide or amino acid sequences, to infer evolutionary relationships between species.
- What we mean with Distance-Based Molecular Data?
- Distance-based molecular data refers to a type of data used in molecular phylogenetics that quantifies the genetic distance between two or more sequences.
The genetic distance is usually calculated by comparing the nucleotide or amino acid sequences of the molecules being studied.
These distances are then used to construct a dendrogram or phylogenetic tree that shows the evolutionary relationships between the sequences. - Distance-based methods are one of two main approaches to molecular phylogenetics, the other being character-based methods.
In contrast to character-based methods, which focus on the presence or absence of specific molecular features, distance-based methods are based solely on the degree of similarity between sequences.
- Distance-based molecular data refers to a type of data used in molecular phylogenetics that quantifies the genetic distance between two or more sequences.
- What are the Differences between Character-Based and Distance-Based Molecular Data?
- In molecular phylogenetics, there are two main types of molecular data: character-based and distance-based.
- Character-based data refers to the actual nucleotide or amino acid sequences that are being compared between different organisms.
These sequences are used to infer the evolutionary relationships between the organisms, and can be analyzed using a variety of methods, such as maximum parsimony, maximum likelihood, and Bayesian inference. - Distance-based data, on the other hand, refers to the calculated distance or dissimilarity between sequences.
This can be done using various distance metrics, such as Jukes-Cantor, Kimura, or p-distance.
These distances are then used to construct a distance matrix, which can be used to infer phylogenetic relationships between organisms using methods such as neighbor-joining or UPGMA.
- Character-based data refers to the actual nucleotide or amino acid sequences that are being compared between different organisms.
- The main difference between the two types of data is that character-based data retains more information about the actual sequences being compared, while distance-based data only retains information about the calculated distances between sequences.
Character-based methods can be more accurate, but they can also be computationally intensive and require more data, while distance-based methods are generally faster and require less data, but may be less accurate.
- In molecular phylogenetics, there are two main types of molecular data: character-based and distance-based.
- What are Phenetics?
- Phenetics is a method of constructing phylogenetic trees based on overall similarity among organisms.
It is also known as numerical taxonomy or similarity-based taxonomy.
In phenetics, the evolutionary relationships among taxa are inferred solely from the overall similarity of their phenotypic or molecular characters, without considering the underlying mechanisms of evolution.
Phenetic methods typically use algorithms to calculate pairwise distances or similarities between taxa based on their characters, and then use these distances to construct a tree using clustering techniques.
While phenetics was popular in the past, it has largely been supplanted by cladistics, which emphasizes the importance of shared derived characters in inferring evolutionary relationships.
- Phenetics is a method of constructing phylogenetic trees based on overall similarity among organisms.
- What is an Operational Taxonomic Unit (OTU)?
- An Operational Taxonomic Unit (OTU) is a unit of classification used in biological classification systems.
It is a term commonly used in molecular biology to define a group of organisms that are similar based on some pre-defined criteria.
These criteria can be based on genetic similarity, morphological characteristics, or ecological similarity. - In molecular biology, OTUs are typically defined based on DNA sequences. Sequences that are similar enough are assigned to the same OTU.
The threshold for similarity is usually set based on the level of sequence divergence that is expected within a particular group of organisms.
For example, a sequence similarity threshold of 97% might be used to define OTUs for bacterial 16S rRNA gene sequences. - OTUs are used to study diversity and relationships among different groups of organisms. They are particularly useful in studies of microbial communities, where traditional taxonomic classification can be difficult due to the large number of species and the difficulty of culturing many of them in the laboratory.
- An Operational Taxonomic Unit (OTU) is a unit of classification used in biological classification systems.
- Who are Cladists, what are they Intrested in?
- Cladists are scientists who study evolutionary relationships among organisms and are interested in reconstructing the evolutionary history of organisms using shared, derived characteristics or traits.
They focus on the identification of monophyletic groups, or clades, which are groups of organisms that share a common ancestor and all of its descendants.
Cladists believe that classification should be based on these monophyletic groups, and they emphasize the importance of using phylogenetic trees to illustrate the evolutionary relationships among organisms.
- Cladists are scientists who study evolutionary relationships among organisms and are interested in reconstructing the evolutionary history of organisms using shared, derived characteristics or traits.
- How can we Reconstruct the Phylogeny?
- The reconstruction of phylogeny can be done using various methods, including:
- Maximum Parsimony (MP) - This method involves selecting the tree that requires the least number of evolutionary changes, or the most “parsimonious” tree.
- Maximum Likelihood (ML) - This method calculates the probability of observing the sequence data given a particular phylogenetic tree.
- Bayesian Inference (BI) - This method involves calculating the probability of different phylogenetic trees given the sequence data, using Bayes’ theorem.
- Neighbor-Joining (NJ) - This method constructs a tree by iteratively joining the most closely related sequences.
- Distance-based Methods - This method constructs a tree based on the pairwise distances between sequences
- Hybrid methods - These methods combine different techniques, such as combining distance-based and character-based approaches.
- Ultimately, the choice of method depends on the type of data available, the evolutionary processes being studied, and the researcher’s preferences.
- The reconstruction of phylogeny can be done using various methods, including:
- What are the Advantages and Disadvantages of Dustance-Based Methods?
- Distance-based methods have some advantages and disadvantages:
- Advantages:
- They are computationally fast and efficient, making them useful for analyzing large datasets.
- They can handle missing data and gaps in the sequence alignments.
- They are relatively easy to interpret and understand.
- They can be used with a variety of distance measures, allowing for flexibility in the analysis.
- They can provide information on the amount of divergence between sequences.
- Disadvantages:
- They assume a constant rate of evolution over time and across all sites, which may not be accurate.
- They do not take into account the evolutionary process and can lead to incorrect tree topologies.
- They can be affected by long-branch attraction, where rapidly evolving lineages are incorrectly grouped together due to the convergence of mutations.
- ==They may not be suitable for closely related taxa==, as the distances between sequences can become too small to accurately estimate.
- They do not incorporate information on the underlying genetic mechanisms, such as recombination or gene conversion.
- Advantages:
- Distance-based methods have some advantages and disadvantages:
- What are Phenograms?
- Phenograms are types of dendrograms or phylogenetic trees that depict the similarities and differences between species or other taxa based on their phenotypic traits.
They are constructed using distance-based methods, which measure the pairwise distance between the taxa and arrange them in a hierarchical manner based on their similarities.
Phenograms are commonly used in fields such as taxonomy, ecology, and evolutionary biology to understand the relationships between organisms based on their physical or observable characteristics.
==However, since they rely on phenotypic data they may not always accurately reflect the true evolutionary relationships between taxa, particularly in cases where convergent evolution or other evolutionary phenomena may lead to similarities between unrelated groups==,.
- Phenograms are types of dendrograms or phylogenetic trees that depict the similarities and differences between species or other taxa based on their phenotypic traits.
—————————————————————
IMPORTANTE
IMPORTANTE I dati molecolari usati per generare alberi filogenetici appartengono a due categorie: Carattere e Distanza. Caratteri: well-defined feartures che accadono in pochi casi.
Distanza: misura della differenza tra due set di data.
IMPORTANTE Phenetics: (character based) si raggruppano unità taxonomiche basandosi sulle loro caratteristiche fisiologiche. #IMPORTANTE Cladisti: alberi filogenetici (distance based) distinguono omologie in: Apomorfismi: omologie recenti, che distinguono chiaramente due unità taxonomiche. Plesiomorfismi: omologie talmente comuni da non caratterizzare un metodo di distinzione.
IMPORTANTE Parsimony-based Methods: ricerca di un albero filogenetico (distance-based) che richeide il minor numero di cambiamenti evolutivi per spiegare le differenze tra due specie. (Not so high computational complexity) #IMPORTANTE Likelihood-based Methods: ricerca di un albero che sia il più probabile possibile rispetto a tutti gli altri. (High computational complexity)
—————————————————————
Slides with Notes

IMPORTANTE I dati molecolari usati per generare alberi filogenetici appartengono a due categorie: Carattere e Distanza. Caratteri: well-defined feartures che accadono in pochi casi.
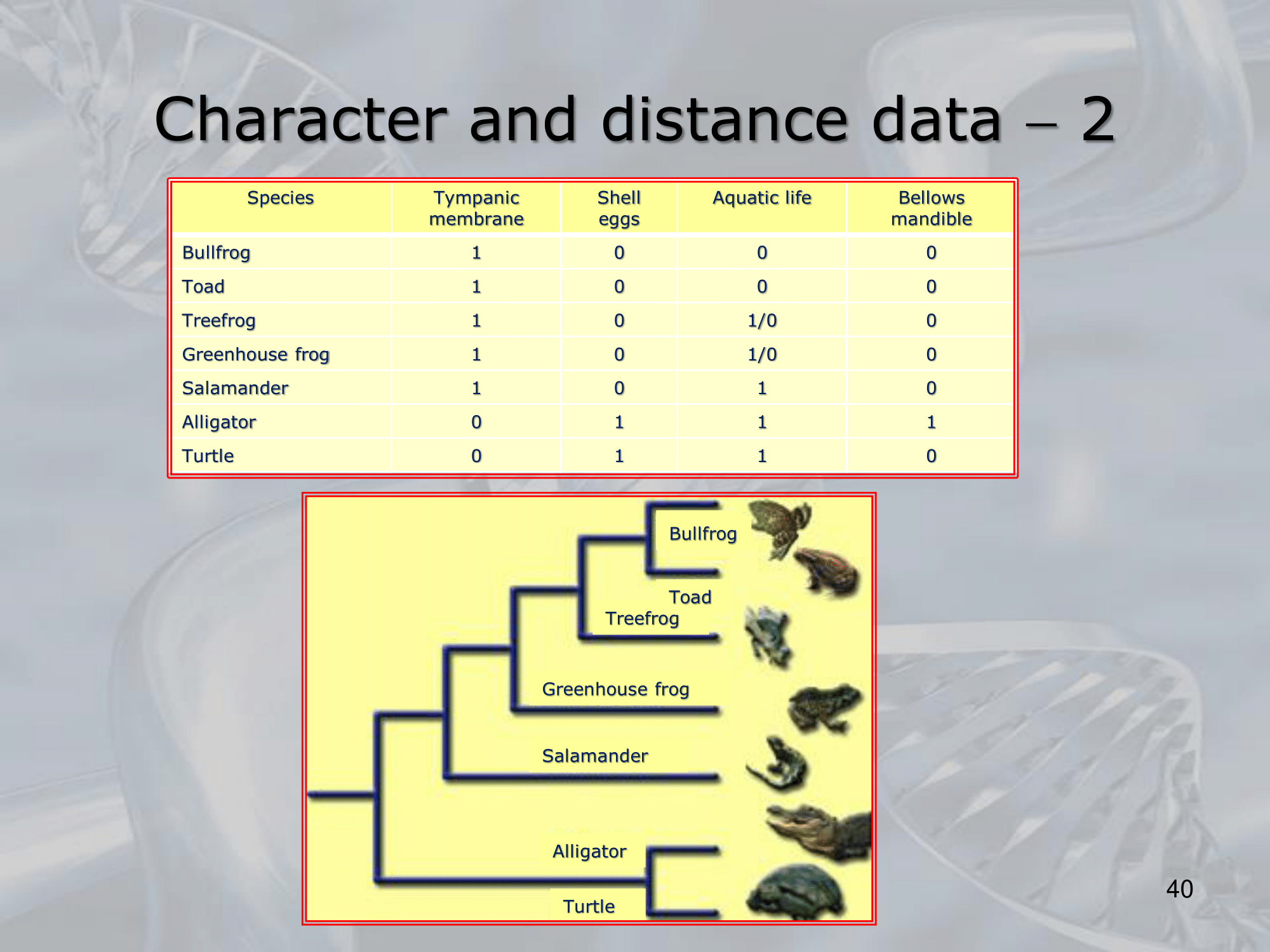







IMPORTANTE Phenetics: (character based) si raggruppano unità taxonomiche basandosi sulle loro caratteristiche fisiologiche. #IMPORTANTE Cladisti: alberi filogenetici (distance based) distinguono omologie in: Apomorfismi: omologie recenti, che distinguono chiaramente due unità taxonomiche. Plesiomorfismi: omologie talmente comuni da non caratterizzare un metodo di distinzione.

IMPORTANTE Parsimony-based Methods: ricerca di un albero filogenetico (distance-based) che richeide il minor numero di cambiamenti evolutivi per spiegare le differenze tra due specie. (Not so high computational complexity) #IMPORTANTE Likelihood-based Methods: ricerca di un albero che sia il più probabile possibile rispetto a tutti gli altri. (High computational complexity)
